This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is Phylogeny?
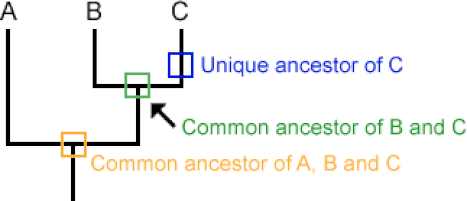
Phylogeny is an estimation of the relatedness of different biological groupings, referred to as taxa. Relationships between taxa are presented in a phylogenic tree. [1]
Phylogenic Trees begin at the root which represents the common ancestor. This is the last ancestor on the tree that all of the other organisms have in common. [2]
This then leads into the branches of the tree. Each branch represents a new taxa. The terminal branches of the trees are unique ancestors which means that there has not been genesis of new taxa from this branch. [2]
Branches are connected at nodes. These represent a common ancestor for the rest of the branches that stem from it and its ancestors. The branches after a node are referred to as a clade. [2]
Phylogenic Trees begin at the root which represents the common ancestor. This is the last ancestor on the tree that all of the other organisms have in common. [2]
This then leads into the branches of the tree. Each branch represents a new taxa. The terminal branches of the trees are unique ancestors which means that there has not been genesis of new taxa from this branch. [2]
Branches are connected at nodes. These represent a common ancestor for the rest of the branches that stem from it and its ancestors. The branches after a node are referred to as a clade. [2]
What Characteristics are Used to Group Taxa?
Physical Features: Phylogenetic studies began by grouping taxa (usually species) according to physical characteristics. This is a fairly simple strategy. For example, snakes and crocodiles both have scales, so they must be more closely related to each other than to a chicken. However, chickens and crocodiles are actually more closely related to each other than to snakes. [3] Thus, appearance is not always a reliable measure of biological relatedness.
Genetics: Taxa can also be grouped according to their genes. Phylogenetics is a technique that determines taxa relationships by comparing sequences of nucleic acids. Phylogenetics can be used to study relatedness of a variety of taxa ranging from a single gene to an entire organism. Genes serve as the blueprints for organisms. Hence, genetic sequences are a good measure of evolution and relatedness. [4]
Proteins: Phylogenic analyses can also be performed using protein relatedness. This technique groups taxa according to their amino acid sequences. This technique is generally used to determine the evolution of a specific protein in several organisms. Proteins are encoded by DNA and carry out most functional activities within an organism. Protein phylogeny is a good measure of evolution as well as functional relatedness of organisms. It can also be helpful in characterizing the molecular evolution of disease. [5]
Genetics: Taxa can also be grouped according to their genes. Phylogenetics is a technique that determines taxa relationships by comparing sequences of nucleic acids. Phylogenetics can be used to study relatedness of a variety of taxa ranging from a single gene to an entire organism. Genes serve as the blueprints for organisms. Hence, genetic sequences are a good measure of evolution and relatedness. [4]
Proteins: Phylogenic analyses can also be performed using protein relatedness. This technique groups taxa according to their amino acid sequences. This technique is generally used to determine the evolution of a specific protein in several organisms. Proteins are encoded by DNA and carry out most functional activities within an organism. Protein phylogeny is a good measure of evolution as well as functional relatedness of organisms. It can also be helpful in characterizing the molecular evolution of disease. [5]
What Methods Are Used to Create Phylogenetic Trees?
|
Neighbor Joining
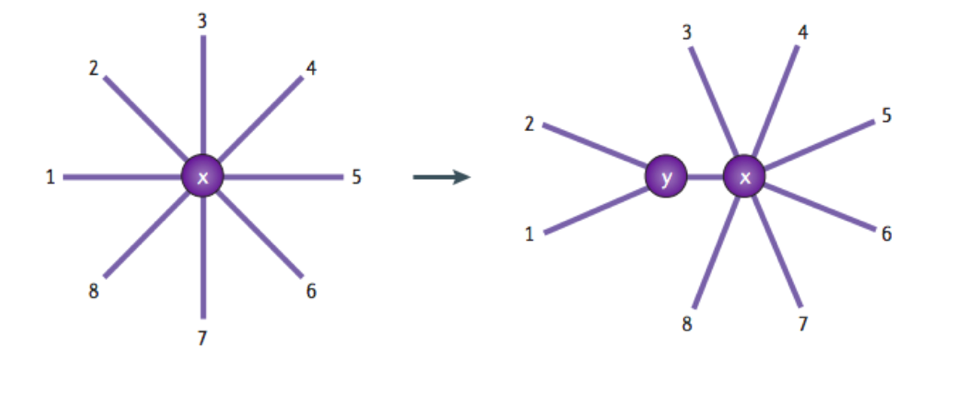
Neighbor joining is a method of building a phylogenetic tree that is based around the branch lengths between nodes. The goal of this method is to construct a tree with the smallest distance between branches. Minimized distance is meant to represent the minimum amount of evolution necessary to achieve the known level of biodiversity that exists today. This method is very fast, but requires excellent sequence alignment. [6] |
|
|
Maximum Likelihood
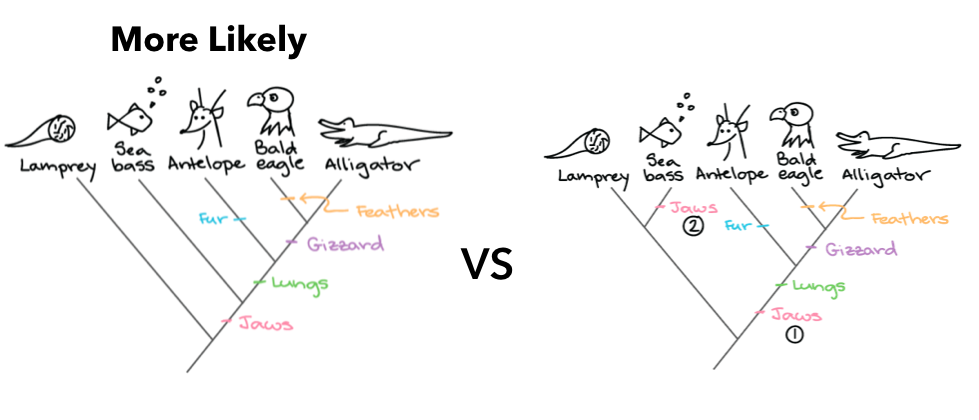
Maximum likelihood is a method of building a phylogenetic tree that is based around minimizing the number of times a unique trait or sequence evolves because it is more likely that there was one evolutionary event that gave rise to a new trait or sequence. For example, it is more likely that jaws evolved once, than that jaws evolved twice independently. This method is easy to see and calculate but may require more assumptions than more qualitative methods. [7] |
How Are Phylogenetic Trees Created?
1. Compile the sequences of interest.
The first step is to gather all of the sequences that will be compared. This may be amino acid sequences or nucleic acid sequences. Once located, the sequences must be compiled into a single document. The example below is a compilation of the amino acid sequences for the MTHFR protein in humans and a variety of model organisms.
The first step is to gather all of the sequences that will be compared. This may be amino acid sequences or nucleic acid sequences. Once located, the sequences must be compiled into a single document. The example below is a compilation of the amino acid sequences for the MTHFR protein in humans and a variety of model organisms.
2. Align the Sequences
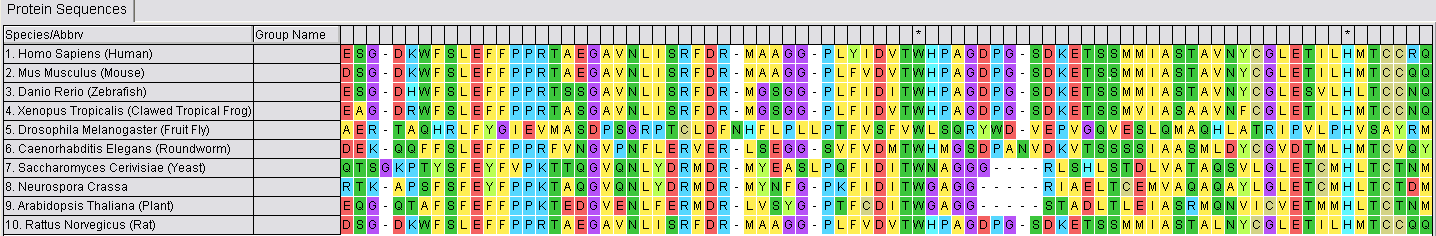
In order to compare the sequences that have been compiled, it is important to line them up with each other. This means matching the parts of the sequences that are the same with each other. This helps to identify which parts of each sequence are unique and aids in identifying when those changes occurred evolutionarily. The example below is a sequence alignment for a segment of MTHFR created using MEGA.
In order to compare the sequences that have been compiled, it is important to line them up with each other. This means matching the parts of the sequences that are the same with each other. This helps to identify which parts of each sequence are unique and aids in identifying when those changes occurred evolutionarily. The example below is a sequence alignment for a segment of MTHFR created using MEGA.
3. Compile the Tree
Once the sequences are aligned, they can be used to compile a phylogenetic tree using any computational method. Examples of trees for MTHFR using the neighbor joining method and the maximum likelihood method are shown below. These trees were also constructed using MEGA.
Once the sequences are aligned, they can be used to compile a phylogenetic tree using any computational method. Examples of trees for MTHFR using the neighbor joining method and the maximum likelihood method are shown below. These trees were also constructed using MEGA.
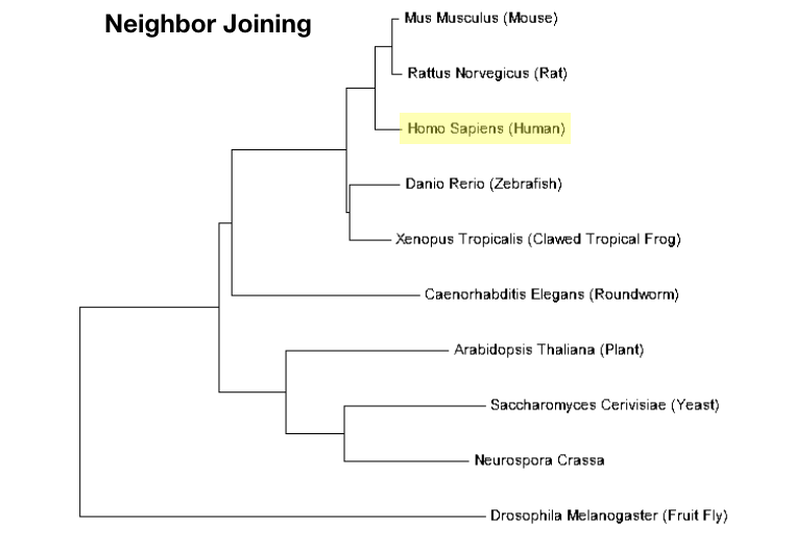
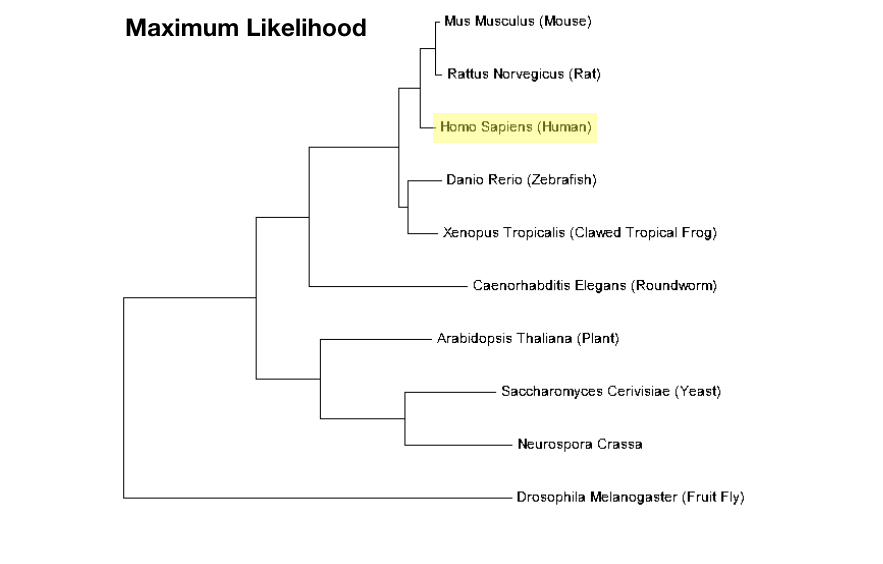
Discussion
Phylogenetic analysis of proteins can help to delineate the evolutionary history of a protein. It can also help us determine how related the proteins present in different species are to each other. Phylogeny can be used to supplement information regarding homology and percent identity, which is a qualitative measurement of the similarity between two protein sequences. From this analysis, it is evident that human MTHFR most related to the form of MTHR in mice and rats and is also closely related to other vertebrate MTHFR orthologs such as zebrafish and frogs. Fruit fly MTHFR seems to belong to an outlying group that is not as closely related to any of the others. All of these results are concordant with homology analyses.
References
[1] Yusav, T., Amena F., Mehvish, F. & Khan, W. (2016). Phylogenetic tree construction of biosurfactant producing organisms. Journal of Global Biostatistics, 5, 4105-4108.
[2] NIH. (n.d.) "Tree Facts: Terminology. NCBI. Retrieved from: https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo7.html
[3] Neil Olander. (n.d). Tree of Life. Tellepallet. Retrieved from: http://abacus.gene.ucl.ac.uk/will/files/TreeOfLife.pdf
[4] University of California at Berkely. (n.d.) Using Trees for Classification. Understanding Evolution. Retrieved from: https://evolution.berkeley.edu/evolibrary/article/phylogenetics_04
[5] Ganfornina, M.D., Gutierrez, G., Bastiani, M. & S.D. (2000). A phylogenetic analysis of the lipocalin protein family. Molecular Biology and Evolution, 17, 114-126.
[6] Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406-425.
[7] Yang, Z. & Rannala B. Molecular phylogenetics: Principles and practice. Nature Reviews Genetics, 13, 303-314.
Images:
Header: https://umr5558-bibiserv.univ-lyon1.fr/flandrois/theme3.html
Image 1: https://evolution.berkeley.edu/evolibrary/article/evo_05
Image 2: https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
Image 3: https://www.khanacademy.org/science/biology/her/tree-of-life/a/building-an-evolutionary-tree
[1] Yusav, T., Amena F., Mehvish, F. & Khan, W. (2016). Phylogenetic tree construction of biosurfactant producing organisms. Journal of Global Biostatistics, 5, 4105-4108.
[2] NIH. (n.d.) "Tree Facts: Terminology. NCBI. Retrieved from: https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo7.html
[3] Neil Olander. (n.d). Tree of Life. Tellepallet. Retrieved from: http://abacus.gene.ucl.ac.uk/will/files/TreeOfLife.pdf
[4] University of California at Berkely. (n.d.) Using Trees for Classification. Understanding Evolution. Retrieved from: https://evolution.berkeley.edu/evolibrary/article/phylogenetics_04
[5] Ganfornina, M.D., Gutierrez, G., Bastiani, M. & S.D. (2000). A phylogenetic analysis of the lipocalin protein family. Molecular Biology and Evolution, 17, 114-126.
[6] Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406-425.
[7] Yang, Z. & Rannala B. Molecular phylogenetics: Principles and practice. Nature Reviews Genetics, 13, 303-314.
Images:
Header: https://umr5558-bibiserv.univ-lyon1.fr/flandrois/theme3.html
Image 1: https://evolution.berkeley.edu/evolibrary/article/evo_05
Image 2: https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
Image 3: https://www.khanacademy.org/science/biology/her/tree-of-life/a/building-an-evolutionary-tree
